来源:科学材料站

多重异质界面强化界面电子转移提升大电流密度下碱性析氢活性
第一作者:任金涛
通讯作者:袁忠勇
通讯单位:南开大学
论文DOI:10.1016/j.apcatb.2024.123817
电催化水分解产生的氢气(H2)被视为一种有吸引力的替代能源,可以取代传统的化石燃料,因为它具有清洁、高效和可再生的特性。目前,基于铂族金属(PGMs)的电催化剂仍然是氢发生反应(HER)的基准催化剂,然而它们的广泛应用受到其有限的供应和高昂的成本的限制。此外,即使是PGMs催化剂,由于H2O解离步骤的固有缓慢性(Volmer反应:H2O + e− + * → H* + OH−),在碱性介质中的活性始终比酸性电解质低2-3个数量级。因此,迫切需要从丰富元素衍生出的高效电催化剂来推动碱性HER的发展。到目前为止,已经广泛合成了包括氧化物、氢氧化物、磷化物、硫化物、碳化物、氮化物及其合金在内的各种过渡金属基电催化剂,并展示出可喜的HER活性。然而,对于大多数催化剂,它们的性能仍然令人不满,并且与商业基准Pt/C相比存在实质性的活性差距。这种性能缺陷源于它们固有的低内在活性和在碱性HER涉及的多个反应步骤中缺乏必要的活性位点。因此,迫切需要开发高效的催化剂合成方法,以制备能够在碱性条件下与Pt相媲美的无PGM催化剂,用于碱性环境中氢气生产。
大量的研究工作致力于开发高效的非PGMs催化剂,其中以3d过渡金属基磷化物作为高效的HER电催化剂引起了相当大的关注,这是由于它们显著的固有活性、优异的导电性以及可调控的结构/组成。最近,通过构建异质结构来优化中间体H*已经成为增强金属磷化物HER活性的关键策略,这主要归因于不同组分之间相互作用和优化的电子结构带来的协同效应。然而,尽管取得了这些进展,大多数基于异质结构的金属磷化物对碱性HER的电催化性能仍然不及PGM催化剂,原因在于其H2O吸附能力差以及在异质结构中H2O解离存在较高的能垒。因此,迫切需要进一步改进磷化物基催化剂异质界面的局部电荷密度并优化电子带结构,以实现高效碱性HER电催化剂的开发。
最近,在基于过渡金属的经济高效催化剂方面取得了显著的突破和实质性进展。为了提高电解效率,工业规模的电解槽通常在20-40 wt% KOH电解液中操作,并在40到80°C的高温条件下工作。这些条件要求使用更为坚固和活性的电极材料,能够经受住严酷环境的考验。具体来说,工业级电极必须提供足够高效的催化位点、快速的电荷和质量传输速率,以及强大的附着力,以承受气泡剧烈生成的挑战。然而,大多数实验室开发的电催化剂不适用于工业应用,主要是由于存在惰性质量传输和活性物质与导电基底之间粘附不足的挑战,尤其是在水分解中以相对较高的电流密度(超过400 mA cm−2)运行时。为了满足工业操作条件的严格要求,工业电催化剂需要具备一些关键属性,包括自支撑的电极结构、丰富的可接触的活性位点、有利的表面形态以促进快速气体生成、卓越的化学和结构稳定性,以及使用易获得的原材料进行经济高效合成的过程。因此,在集流体上集成活性异质结构的直接制备策略,作为自支撑电极,以最大程度发挥结构和组成的优势,对于在更大的电流密度下实现快速的电化学制氢,特别是在工业环境中,是至关重要的。
针对这些挑战,南开大学袁忠勇课题组采用密度泛函理论(DFT)预测,以确定Ni2P-NiSe2异质界面的局部电荷密度和电子带结构在与MoO2基底耦合时经历显著优化,从而改善H2O吸附,降低H2O解离的能垒,并促进H*的吸附/脱附,共同加速碱性HER的反应动力学。在这些理论的指导下,借助一种直观且可扩展的策略,通过将界面丰富的Ni2P-NiSe2异质结构牢固地锚定在镍泡沫上的无定形MoOx纳米棒上(Ni2P-NiSe2/MoOx/NF)作为自支撑电极。由于其多个活性位点和自支撑结构的协同耦合相互作用以及增强的固有活性,所得的Ni2P-NiSe2/MoOx/NF催化剂展现出卓越的碱性HER性能。值得注意的是,Ni2P-NiSe2/MoOx/NF催化剂在超低过电位263和424 mV下分别实现了500和1000 mA cm−2的电流密度,并表现出卓越的稳定性,甚至在1.0 M KOH中持续运行了200小时。由于其多个活性位点和自支撑结构,该催化剂还表现出优异的氧进化反应(OER)活性和稳定性。因此,Ni2P-NiSe2/MoOx/NF催化剂在水碱电解中具有巨大的潜力,特别是在与光伏电池集成以实现太阳能驱动的水电解时,可在环境条件下实现高达26.17 μL s−1的气体产生速率。此外,该催化剂在工业条件下(30% KOH,65°C)在高电流密度下表现出稳定的性能。最后,在阴离子交换膜(AEM)水电解槽中使用时,该催化剂在细胞电压为2.7 V时实现了1.3 A cm−2的较大电流密度,并具有持久的稳定性。

图1. DFT理论计算结果。
为了理解Ni2P和NiSe2(Ni2P-NiSe2)之间的异质界面及其与MoO2(Ni2P-NiSe2/MoO2)的协同耦合的影响,我们进行密度泛函理论(DFT)计算。在Ni2P-NiSe2界面处,局部电荷密度明显增加,表明强大的界面电子相互作用。图1c表明,Ni2P和NiSe2的εd值分别为-1.69和-1.34 eV,表明它们对反应中间体的吸附特性要么过弱要么过强。相反,Ni2P-NiSe2的εd值为-1.49 eV,这适合优化氢和氧中间体的吸附,从而提高电催化活性。计算得到的Ni2P-NiSe2的H2O吸附自由能(ΔGH2O),如图1d所示,为-0.71 eV。这个值明显低于单独的Ni2P和NiSe2组分的值,强调了异质界面工程有效促进了H2O的吸附。这种H2O吸附的增强反过来促进了反应物的表面作用,从而加速了后续的H2O解离步骤。如图1e所示,Ni2P-NiSe2界面上P位点的ΔGH*值(-0.31 eV)与单独的Ni2P(-0.43 eV)和NiSe2(-0.59 eV)组分相比显著接近最佳值。
这一观察强调了Ni2P和NiSe2之间的界面相互作用优化了氢的吸附,从而提高了内在HER活性。此外,我们计算了H2O解离的能垒,如图1f所示。Ni2P、NiSe2和Ni2P-NiSe2的H2O解离能垒分别为0.54、0.63和0.40 eV。这种能量差异表明Ni2P-NiSe2界面的构建主要促进HO-H键的解离,从而增加活性位点上H*的浓度。因此,这种增强显著加速了Ni2P-NiSe2界面上的碱性HER动力学。
另一方面,这些理论计算的发现使我们想知道是否可以进一步优化Ni2P-NiSe2的中间体吸附能和反应能垒,以获得卓越的性能。为此,我们通过将Ni2P-NiSe2锚定到MoO2基底上(Ni2P-NiSe2/MoO2)引入了一个富含界面的体系,以进一步微调电子结构并创建额外的活性位点。引入MoO2导致ΔGH2O值从Ni2P-NiSe2的-0.71 eV变为-0.86 eV(图1d),表明电子不足的Mo位点展现出对水吸附有利的能级。此外,Ni2P-NiSe2/MoO2在P位点的ΔGH*值为-0.05 eV(图1e),低于Ni2P-NiSe2的值,暗示着MoO2引入后吸附行为更为优化。最值得注意的是,在Ni2P-NiSe2/MoO2结构中,H2O解离的能垒降至0.16 eV(图1f),表明MoO2与Ni2P-NiSe2界面的耦合进一步促进了HO-H键的解离。在Ni2P-NiSe2/MoO2结构中,带正电的MoO2表面促使富电子的氧原子吸附在H2O中,激活H2O分子,使其更易于在MoO2上发生解裂。MoO2和Ni2P-NiSe2界面之间的这种协同效应有助于整体性能的提升。
总的来说,在这个多组分的Ni2P-NiSe2/MoO2中,Ni2P-NiSe2和MoO2之间的电子相互作用可以协同促进H2O的吸附,加速H2O的解离,并有效地吸附产生的H*,从而显著促进碱性HER过程中的每个反应步骤,这是根据DFT计算得出的。

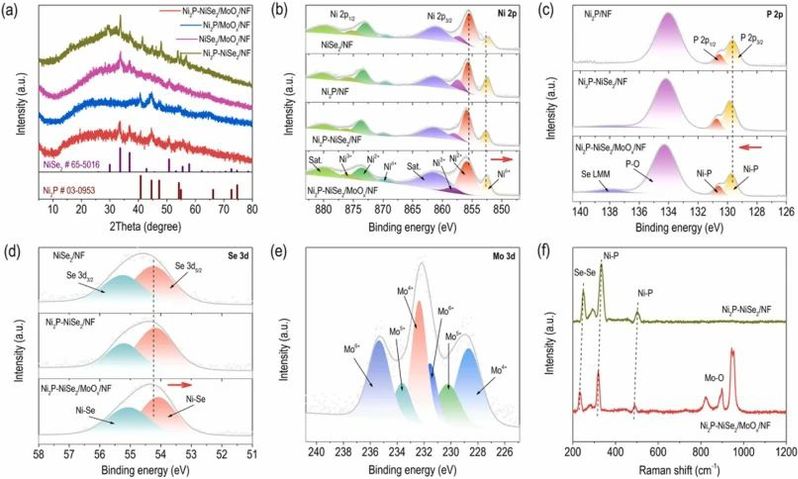
图3. 催化剂的XRD,XPS,和Raman表征。
对于Ni2P-NiSe2/MoOx/NF,XRD图谱(图3a)表明了Ni2P和NiSe2的生成。值得注意的是,在所有制备的样品中,均未观察到与钼基成分有关的可辨认衍射峰,这与它们的TEM观察一致,因此为制备的Ni2P-NiSe2/MoOx异质结构的结晶-非晶性特性提供了互补证据。通过XPS和Raman对催化剂的表面化学组成和电子相互作用进行了研究。相关测试结果如图3b-f所示。比较Ni2P-NiSe2/MoOx/NF和Ni2P-NiSe2/NF中的Niδ+峰的发现,Ni2P-NiSe2/MoOx/NF的结合能出现了轻微的负偏移,表明在界面Ni原子处电子密度的富集。这个偏移可能是由于MoOx基底向Ni2P-NiSe2的电子转移引起的,进一步突显了异质结构内复杂的电子相互作用。因此,与理论预测一致,这有望共同发挥作用并获得卓越的碱性析氢催化活性。
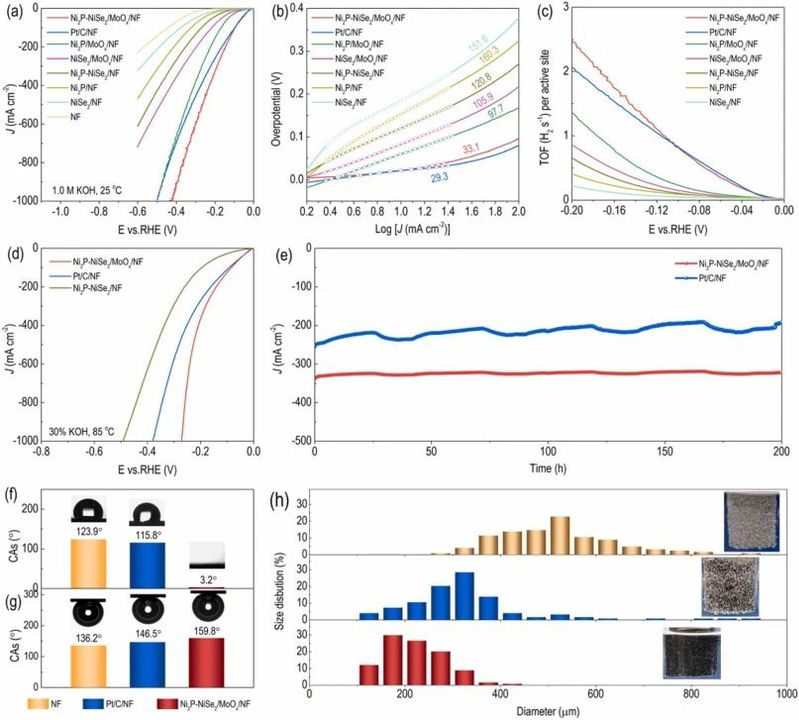
受有Ni2P-NiSe2/MoOx/NF催化剂的复杂界面结构的启发,我们系统评估了其HER的电催化性能。图4a中的iR校正的极化曲线明确展示了异质结构工程后电催化剂的显著提高的HER活性。值得注意的是,Ni2P-NiSe2/NF催化剂需要119、271和428 mV的过电位,分别在10、100和500 mA cm-2的电流密度下。然而,在与MoOx基底耦合后,得到的Ni2P-NiSe2/MoOx/NF在10、100和500 mA cm-2时只需23、96和263 mV的过电位。此外,为了达到工业上有意义的电流密度1000 mA cm-2,Ni2P-NiSe2/MoOx/NF需要424 mV的过电位,低于Pt/C/NF(501 mV),显示了该催化剂在工业相关应用中的实质性潜力。如图4b中Tafel图所示,Ni2P-NiSe2/MoOx/NF表现出最低的Tafel斜率为33.1 mV dec-1,相较于其他催化剂,表明其通过Volmer-Heyrovsky机制显著改善的HER动力学。在200 mV的过电位下,Ni2P-NiSe2/MoOx/NF实现了2.47 s-1的TOF(图4c),这比Pt/C/NF以及其他样品都大,表明了更优越的内在活性。受到卓的HER性能的鼓舞,Ni2P-NiSe2/MoOx/NF在工业条件(30% KOH,85 ℃)中进一步进行测试,以评估其实际适用性。在这种苛刻的条件下,相较于Ni2P-NiSe2/NF(-0.197、-0.366和-0.489 V)和Pt/C/NF(-0.091、-0.285和-0.381 V)较大的过电位,Ni2P-NiSe2/MoOx/NF仅需要较低的电位,即-0.082、-0.227和-0.271 V,就能提供100、500和1000 mA cm-2的阴极电流密度(图4d)。
这种活性表现验证了Ni2P-NiSe2/MoOx/NF在实际应用中的显著潜力和高效性。显著的是,如图4e所示,经过在-0.20 V vs. RHE电位下进行的计时电流测试,Ni2P-NiSe2/MoOx/NF在200小时内表现出卓越的稳定性,始终提供大约-330 mA cm-2的大电流密度。相反,贵金属Pt/C/NF催化剂在相同条件下表现出明显的电流下降。
电催化剂的电化学性能受其质量传递能力的强烈影响,而这可以通过它们的润湿性来评估。为了定量分析各种催化剂之间的润湿性差异,进行了接触角(CA)测量。图4f显示,Ni2P-NiSe2/MoOx/NF在3.2°的水接触角(CA)上表现出最小,与Pt/C/NF(115.8°)和裸露的NF(123.9°)形成对比,表明Ni2P-NiSe2/MoOx/NF具有显著的亲水性。此外,Ni2P-NiSe2/MoOx/NF的气泡CA(图4g)为159.8°,大于Pt/C/NF(146.5°)和裸露的镍网(NF)(136.2°),表明Ni2P-NiSe2/MoOx/NF具有更高的疏水性。这些综合结果表明Ni2P-NiSe2/MoOx/NF具有高效的质量传递和在表面释放气泡的能力。如图4h所示,气泡牢牢附着在裸露的NF和Pt/C/NF表面,迅速增长到300至800 μm的大尺寸范围。相比之下,对于Ni2P-NiSe2/MoOx/NF,小尺寸的气泡(≤300 μm)能够轻松地从表面逃逸到溶液中。根据固液气界面理论,粗糙的电极表面有利于形成不连续的三相界面接触,从而显着减少气泡和电极表面之间的接触区域,从而有利于反应物和气态产物的吸附和解吸。
考虑到Ni2P-NiSe2/MoOx/NF异质结构催化剂中的多重活性组分,我们对其氧还原反应(OER)活性也进行了评估。令人印象深刻的是,Ni2P-NiSe2/MoOx/NF表现出卓越的OER活性,为实现电流密度为10 mA cm−2和500 mA cm−2分别需要241 mV和592 mV的过电位。当测试条件改变为30% KOH、85℃时,实现500 mA cm−2和1000 mA cm−2所需的阳极电位仅分别为351 mV和403 mV,表明了加速的反应动力学。经过OER稳定性测试后,XPS结果显示P和Se几乎消失,而表面氧和金属-氧键合显著增加。EDX图谱进一步表明Ni2P-NiSe2/MoOx的相变为氧化镍水合(氧)氧化物。同时,纳米棒阵列仍然保持不变,尽管纳米棒表面变得粗糙,并且一些纳米片包覆在纳米棒表面。在OER过程中,Ni2P-NiSe2/MoOx/NF从最初的氧化中形成的原位转化的镍水合(氧)氧化物可能是碱性OER的真正活性物质,类似于先前报道的先例,从而导致其卓越的OER活性和稳定性。
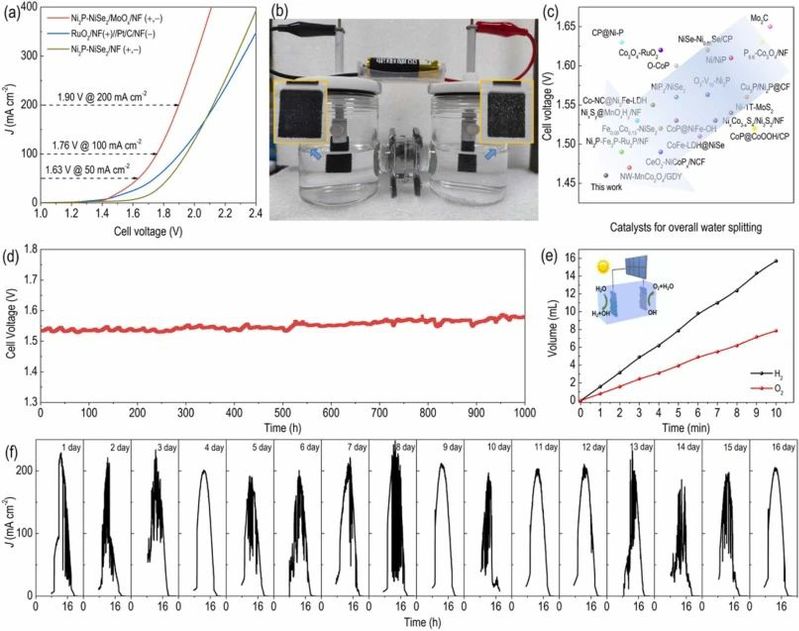
图5. 催化剂的全解水性能。
受Ni2P-NiSe2/MoOx/NF在碱性介质中进行HER和OER中卓越性能的启发,我们组装了一个水分解电解槽,使用Ni2P-NiSe2/MoOx/NF作为双功能电极,采用1.0 M KOH电解质。如图5a所示,Ni2P-NiSe2/MoOx/NF的整体水电解极化曲线在50、100和200 mA cm−2时表现出极低的电池电压,分别为1.63、1.76和1.90 V,优于RuO2/NF||Pt/C/NF(1.73、1.90和2.14 V)和Ni2P-NiSe2/NF(1.83、1.95和2.15 V)。此外,图5b中的照片表明,一节工作电压为1.5 V的单个商业碱性电池可以驱动水电解槽产生可观的气体产量,证实了其出色的电能转化为氢气的效率。此外,根据图5d所示的20 mA cm−2电流密度下的稳态电化学分析,Ni2P-NiSe2/MoOx/NF在持续1000小时以上仍保持着其非凡的活性,符合商业应用的严格要求。通过将水电解槽与商业可用的硅太阳能电池集成,进一步展示了使用可再生电力产生无碳排放的绿色氢气的可行性。以太阳能驱动电力为能源,双功能的Ni2P-NiSe2/MoOx/NF电极迅速催化水分解,并持续释放气泡。为了评估在室外环境中集成系统的性能,以太阳能为能源的水分解单元在自然光照条件下进行了电流时间实验。如图5f所示,所测得的电流密度呈现出与光强变化相关的同步波动,最高电流密度始终在中午获得。
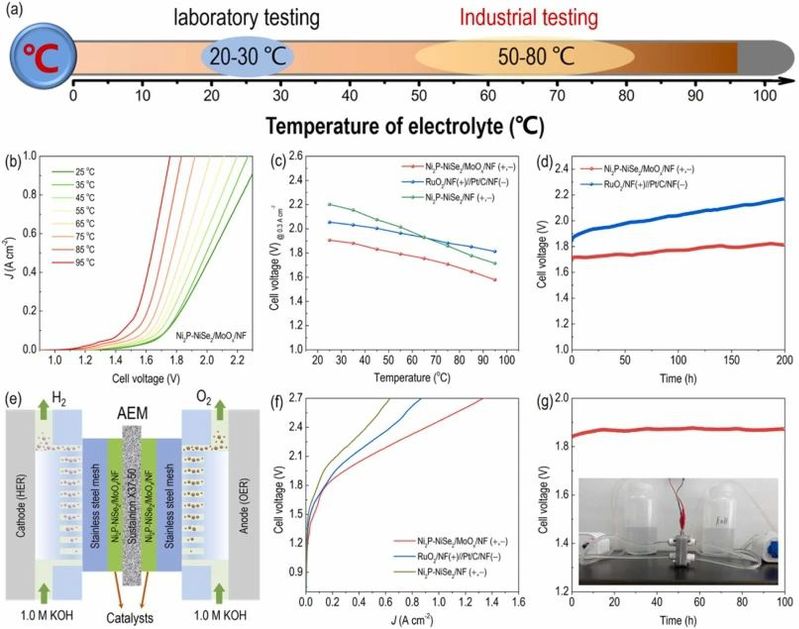
图6. 工业模拟条件下催化剂的产氢性能考察。
在工业水电解系统中,操作温度通常在50到90°C左右,旨在进一步降低水分解所需的整体电压。可以在工业温度(50–90°C)下评估水电解槽的电化学性能,以弥合实验室测试和工业应用之间的差距(图6a)。在一系列温度下测量了Ni2P-NiSe2/MoOx/NF的整体水电解极化曲线。如图6b所示,随着碱性电解液的温度逐渐升高,Ni2P-NiSe2/MoOx/NF电极在相同电流密度下分解水所需的电压降低。当电流密度设定为300 mA cm−2时(图6c),Ni2P-NiSe2/MoOx/NF的水分解电压始终低于RuO2/NF(+)||Pt/C/NF(-)和Ni2P-NiSe2Ni2P-NiSe2/NF在不同温度下的电压。在200 mA cm−2和65°C下对Ni2P-NiSe2/MoOx/NF进行电化学稳定性评估。如图6d所示,Ni2P-NiSe2/MoOx/NF的整体水电解电压衰减速度较慢(0.58 mV h−1),相比之下,RuO2/NF(+)||Pt/C/NF(-)(1.5 mV h−1)的电压衰减速度更快。这表明了Ni2P-NiSe2/MoOx/NF在高温和高电流密度下的卓越运行耐久性,进一步突显了其在工业应用中的潜力。
除了上述评估之外,还构建了一种碱性阴离子交换膜(AEM)电解槽,以评估Ni2P-NiSe2/MoOx/NF在工业条件下的潜在应用。图6e详细描述了组装的AEM水电解槽的结构。使用Ni2P-NiSe2/MoOx/NF作为催化剂的AEM电解槽在一系列电流密度下显示出比RuO2/NF(+)||Pt/C/NF(-)和Ni2P-NiSe2/NF更低的过电位(图6f),并在过渡性碱性水电解槽(AWE)中也是如此。AEM电解槽的改进性能归因于AEM中较低的电荷转移电阻相比AWE电解槽。此外,组装的AEM电解槽在200 mA cm−2下展现了100小时的显著稳定性(图6g)。基于这些结果,Ni2P-NiSe2/MoOx/NF被认为是工业应用中大规模氢气生产的有希望的候选者。
Ni2P-NiSe2/MoOx/NF催化剂表现出的卓越催化性能可以归因于几个关键因素:(i) 多重界面之间的复杂协同作用在电子结构的修改中发挥了关键作用,导致对反应中间体的优化吸附能力。(ii) 由Ni2P-NiSe2/MoOx/NF异质结构组成的电极显著促进了这些异质结构与底层镍泡沫基底之间的界面电子传输。这种界面电子传输的增强对活性物种的质量活性和固有活性起到了支撑作用。(iii) 坚固的催化剂-基底相互作用和降低的金属溶解率显著有助于提高Ni2P-NiSe2/MoOx/NF所表现出的卓越稳定性,确保其在长时间操作期间保持持续的催化性能。(iv) Ni2P-NiSe2/MoOx/NF纳米棒阵列的纳米工程作为一种有效的缓解策略,有助于减轻电催化过程中与气泡粘附相关的不利影响。
袁忠勇教授 beat365中国在线体育教授、博士生导师。南开大学新催化材料科学研究所所长。英国皇家化学会会士。兼任国际期刊《RSC Advances》副主编,《Advanced Materials Science and Technology》主编,以及《精细石油化工》、《无机盐工业》、《Current Catalysis》、《Journal of Engineering》等期刊编委,《催化学报》、《Frontiers of Chemical Science and Engineering》、《石油学报(石油加工)》等期刊客座编辑。已在Chem. Soc. Rev.,Angew. Chem. Int. Ed.,Adv. Mater.,Adv. Energy Mater.,Adv. Funct. Mater.,ACS Nano等重要期刊上发表SCI 收录论文420余篇,论文已被他人引用20000余次,H-index为72。出版英文专著1部和专著章节5篇,获中国发明专利授权10余项。
研究团队介绍:
课题组主要从事非贵金属纳米材料的合成及其能源催化应用,具有充足的科研经费、实验室空间和全新的实验设备,欢迎报考硕士、博士,申请联培研究生、科研助理和博士后。

